 027-62435310 |
027-62435310 |
 service@vikimarina.com |
service@vikimarina.com |
Smart细菌完成图——更准确、更快速、更经济!
发布时间:2020-05-14 15:29 作者:龙8总区科技
龙8总区基因最新推出的 Smart 细菌完成图是高准确和经济的完美结合,是细菌完成图的首选。随着测序技术的不断发展,细菌基因组完成图组装经历了三个大的阶段:第一阶段,准确+昂贵;第二阶段,经济+准确度较高;第三阶段,准确+经济+快速。
将细菌基因组测序从头组装,得到一条连续完整的基因组序列(1 contig,0 gap)就是细菌完成图。再次在此基础上进行基因组组分、基因功能注释和比较基因组等分析。
在高通量测序技术发展的时代,根据测序技术的个性特性,细菌完成图组装经历了三个个大的阶段。
第一阶段,高通量测序+Sanger测序——不惜一切代价完成
主要使用的是 illuminaIllumina 和454测序技术,构建小片段文库( 二代illumina一般是500bp,由于454已经不再使用就不作讨论)组装得到contig,再使用mate-pair文库(2Kb、5K或10K等)把contig连接成scaffold,最后使用sanger测序进行补洞。完全是一个体力活加上烧钱的活,费用一般在几十万。
在这里需要科普一下mate-pair文库,在长度长的时代已经不多见了,只有骨灰级的生信息分析人使用过。首先将基因组DNA随机打断到特定大小(2-10 kb)的片段,生物素标记后环化,在把环化的DNA打断成400-600 bp的片段,富集生物素标记的片段。其实就是测长片段末端的序列,然后把contig连起来,类似于测BAC末端。
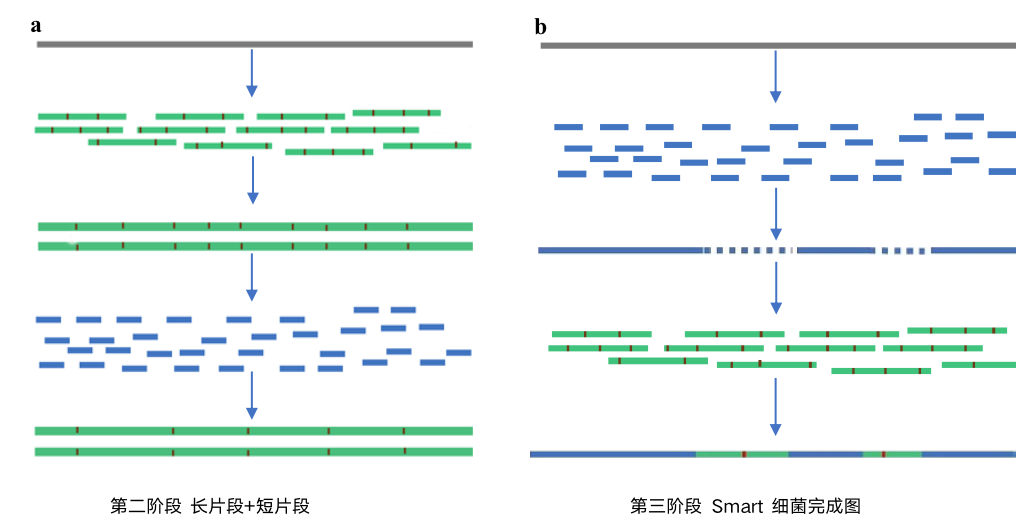
第二阶段 长片段+短片段——低成本
PacBio的长reads的出现使细菌完成图组组装变得不再费时费力,细菌基因组完成图进入了第二个阶段。PacBio平台单碱基错误率在15%左右(现在在10%左右),组转装完成后使用二代illumina数据进行纠错,的得到准确度高的基因组完成图。
随后Nanopore测序的出现,读长更长,加上建库和测序成本降低,细菌基因组完成图成本进一步降低。缺点和PacBio平台一样,开始错误率15%左右,现在错误率在10%左右,组装完成后使用短序列去纠错买得到较为准确的完成图。
1 先讲讲PacBio平台,测序错误主要以为插入和缺失为主,插入占的比例更高,组装基因组的时候为了的得到更长的序列,使用IsoCLR测序模式,序列是不能进行自我矫正的(短序列CCS序列可以进行自我矫正)。由于细菌基因组没有内含子结构,且有多数顺反子结构存在,这种测序错误会引入大量的移码突变,对基因功能的注释和后续的研究影响较大。
2 再讲讲Nanopore平台,测序错误主要是单碱基突变,这一类型的错误率容易解决,经过自纠错和二代illumina数据进行纠错,这种错误率可以降低到0.01%到0.03%。还有一类错误是在序列中出现多个连续单碱基的时候,会引入插入缺失突变,并且这种突变很难进行自纠错,是只能使用二代illumina数据进行纠错。不过这一类序列在细菌中出现得非常少,对基因功能得注释的影响可以忽略。
在这一阶段,费用降下来了,已经是意见一件非常了不起的事情了。组装的错误率也在不断的程序员的不断努力下有所改善,不过还是不能达到理想的状态。
第三阶段, Smart 细菌完成图
“穷则思变,变则通,通则达”,既然仪器上的改进是有限的,那么只能从分析策略的角度去解决问题。
龙8总区基因独立开发的基于Nanopore和Illumina二代测序联合组装的全新技术巧妙应用Illumina二代测序数据高准确度和Nanopore长度长的优点,规避长序列错误率高的问题。细菌完成图变得更快,更经济,当然更准确,进入Smart 细菌完成图时代。
Smart 细菌完成图其实很简单,先用高准确度的二代Ilumina数据进行组装,得到高质量的Contig,然后用Nanopore数据将contig连接成完成图,然最后再把用二代回去数据进行纠错。成功解决Nanopore序列质量低和二代 Ilumina短序列组装难的问题,得到高质量的细菌基因组完成图。该策略由二代 Ilumina数据的决定准确度,同时可规避Nanopore 数据拆分错误引入的序列污染,快速的得到高质量的细菌基因组完成图,错误率降低到0.001%级别。
 这些都在龙8总区基因推出的Smart 细菌完成图中
这些都在龙8总区基因推出的Smart 细菌完成图中的得到完美的解决。更准确,更快,更经济。
将细菌基因组测序从头组装,得到一条连续完整的基因组序列(1 contig,0 gap)就是细菌完成图。
在高通量测序技术发展的时代,根据测序技术的
第一阶段,高通量测序+Sanger测序——不惜一切代价
主要使用的是 illuminaIllumina 和454测序技术,构建小片段文库( 二代illumina一般是500bp,由于454已经不再使用就不作讨论)组装得到contig,再使用mate-pair文库(2Kb、5K或10K等)把contig连接成scaffold,最后使用sanger测序进行补洞。完全是一个体力活加上烧钱的活,费用一般在几十万。
在这里需要科普一下mate-pair文库,在长度长的时代已经不多见了,只有骨灰级的生信息分析人使用过。首先将基因组DNA随机打断到特定大小(2-10 kb)的片段,生物素标记后环化,在把环化的DNA打断成400-600 bp的片段,富集生物素标记的片段。其实就是测长片段末端的序列,然后把contig连起来,类似于测BAC末端。
第二阶段 长片段+短片段——低成本
PacBio的长reads的出现使细菌完成图
随后Nanopore测序的出现,读长更长,加上建库和测序成本降低,细菌基因组完成图成本进一步降低。缺点和PacBio平台一样,开始错误率15%左右,现在错误率在10%左右,组装完成后使用短序列去纠错
1 先讲讲PacBio平台,测序错误主要以
2 再讲讲Nanopore平台,测序错误主要是单碱基突变,这一类型的错误率容易解决,经过自纠错和二代illumina数据进行纠错,这种错误率可以降低到0.01%到0.03%。还有一类错误是在序列中出现多个连续单碱基的时候,会引入插入缺失突变,并且这种突变很难进行自纠错,
在这一阶段,费用降下来了,已经是
第三阶段, Smart 细菌完成图
“穷则思变,变则通,通则达”,既然仪器上的改进是有限的,那么只能从分析策略的角度去解决问题。
龙8总区基因独立开发的基于Nanopore和Illumina二代测序联合组装的全新技术巧妙应用Illumina二代测序数据高准确度和Nanopore长度长的优点,规避长序列错误率高的问题。细菌完成图变得更快,更经济,当然更准确,进入Smart 细菌完成图时代。
Smart 细菌完成图其实很简单,先用高准确度的二代Ilumina数据进行组装,得到高质量的Contig,然后用Nanopore数据将contig连接成完成图,
上一篇:没有了
下一篇:最新生物医学类期刊2020年SCI影响因子预测查询


