027-62435310 |
027-62435310 |
 service@vikimarina.com |
service@vikimarina.com |
一文了解MetaHi-C应用方向
上期给大家介绍了宏基因组分箱方法及Hi-C在宏基因组中的优势,总的来说宏基因组Hi-C不仅可以辅助分箱得到更多高质量的MAGs,实现复杂环境样本中的新物种发现;同时也可以界定环境样品中微生物与质粒和病毒之间的宿主关系,研究复杂微生物组样本中诸如抗生素抗性基因等的宿主来源。
本期我们将通过两篇MetaHi-C文章,详细了解它的实际应用。

英文标题:ViralCC retrieves complete viral genomes and virus-host pairs from metagenomic Hi-C data
发表期刊:Nature Communications
影响因子:16.6
发表时间:2023年1月31日
作者开发了一款针对病毒的工具ViralCC,通过宏基因组Hi-C数据,从环境样品中恢复了高质量病毒基因组及检测病毒-宿主对,并使用多个不同微生物生态系统(如人类肠道、牛粪和废水)的模拟和真实宏基因组Hi-C数据集,发现该工具优于现有基于Hi-C的分箱工具以及专门用于宏基因组病毒分箱的其他工具。工作流程如下图:
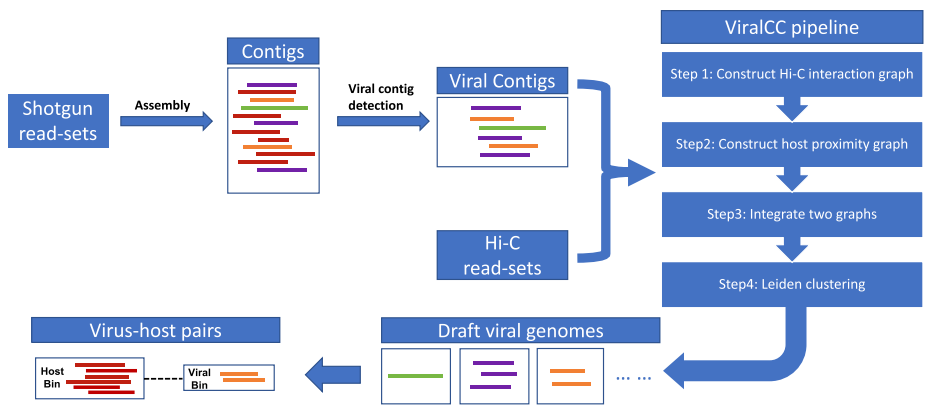
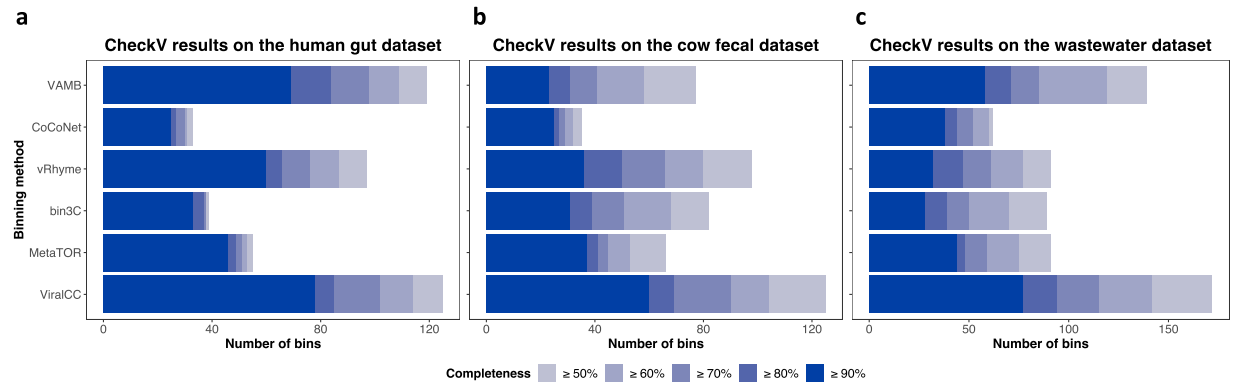
分别从人肠道、牛粪便和废水样本中检测到791、1338和2757个病毒contigs,通过分箱分析后,分别得到了465、574和1240个病毒MAGs(vMAGs),大小从3,001 bp到461,626 bp不等. 将ViralCC与多种方法进行比较,表明ViralCC可以从三个真实环境样本中组装得到更多完整的vMAG,且vMAG总数也最高。
通过对vMAGs的注释,发现vMAGs以尾状病毒目噬菌体为主,分别属于肌病毒科、虹膜病毒科和足病毒科;对于非病毒的contigs,通过Hi-C分箱共得到1253 MAGs,优势物种为Burkholderiales、Pseudomonadales等;基于Hi-C的互作信息,发现Myoviridae的vMAGs主要针对Burkholderiales为目的宿主,Siphoviridae的vMAGs可感染拟杆菌科细菌。
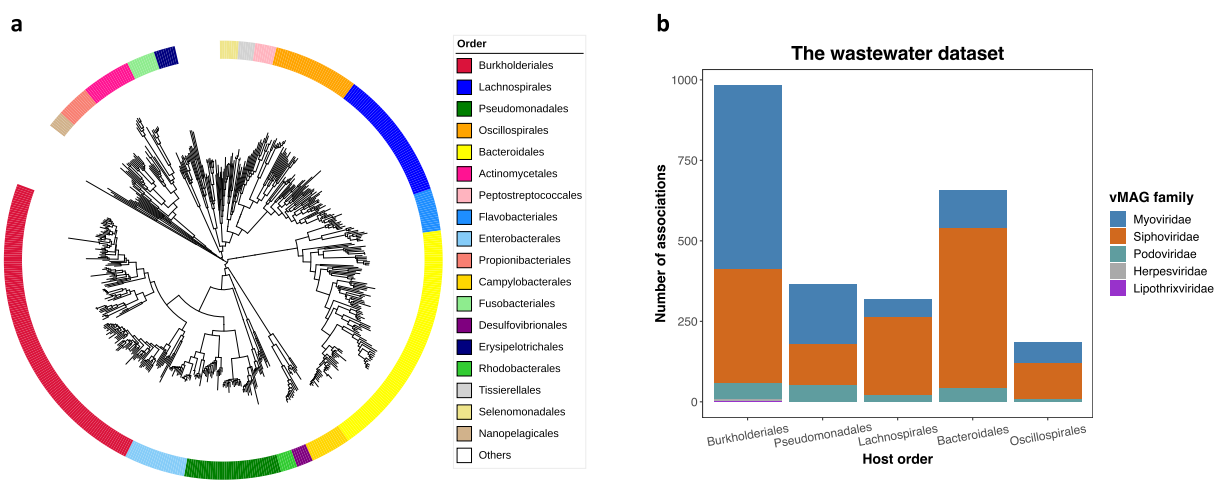
注:图a为MAGs的物种进化树;图b为vMAGs与宿主的对应关系

英文标题:Genome-centric analysis of short and long read metagenomes reveals uncharacterized microbiome diversity in Southeast Asians
发表期刊:Nature Communications
影响因子:16.6
发表时间:2022年10月13日
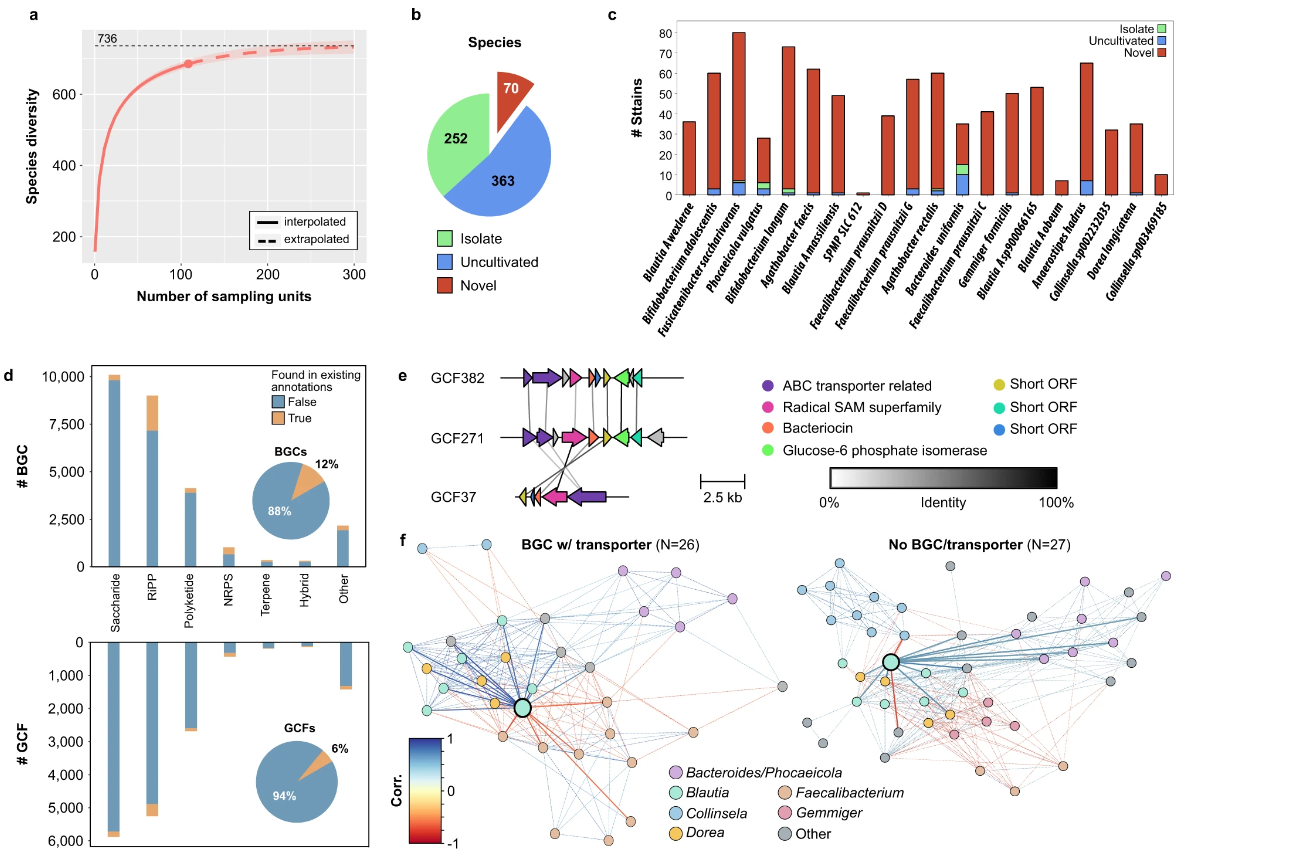
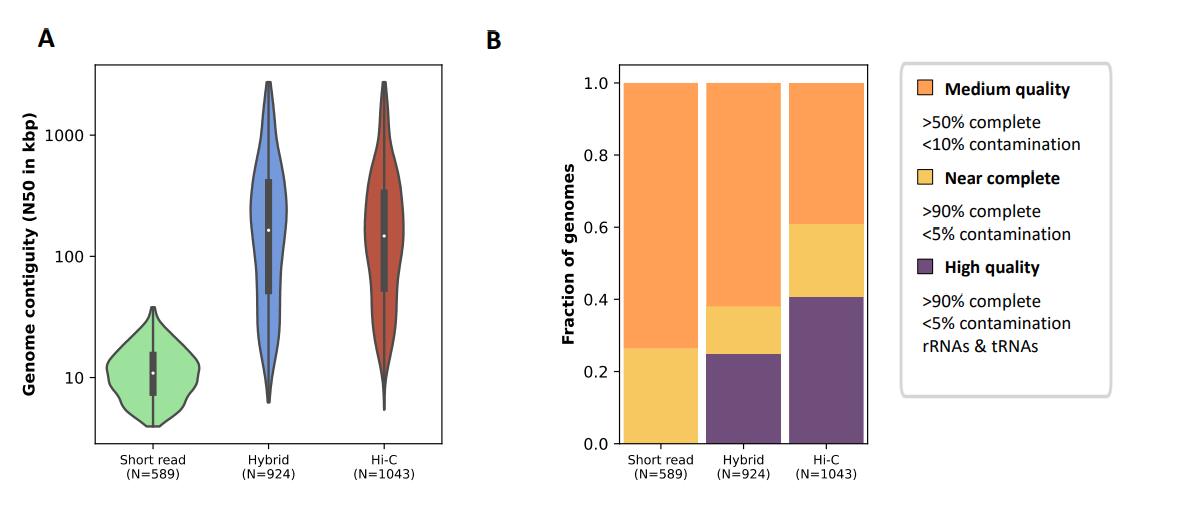
研究发现人类肠道微生物组与各种癌症、代谢、免疫学和神经疾病相关。宏基因组测序可用于解析微生物的群落结构、物种组成、系统进化、基因功能和代谢网络等。本研究利用Illumina与ONT测序平台结合Hi-C技术,对来自新加坡三个种族的109个人群肠道微生物组进行了深入表征,全面重建了4497个中等和高质量的MAGs。
主要结果
1. 构建了特定人群的高质量肠道微生物参考目录
研究发现,加入长读长测序数据,可以显著改善基因组组装的连续性; 109个样品的混合组装获得了4497个MAGs,使用短读长仅获得了2789个MAGs。混合组装获得了更高质量的基因组,获得了短读长测序组装没有得到的MAGs。最后结合了Hi-C数据,增加了获得高质量MAG的比例,并使完整的基因组比例增加了一倍。
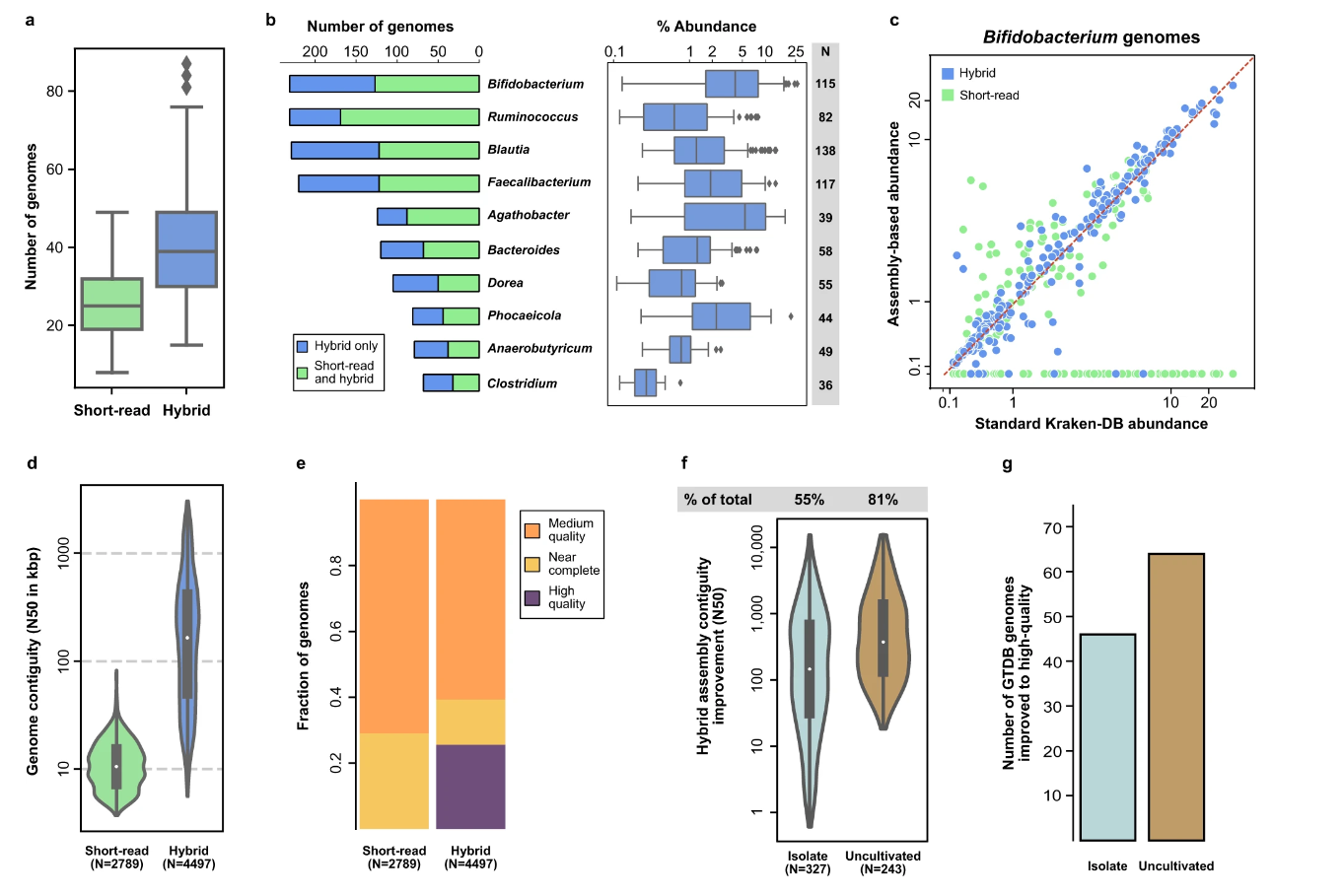
2. 亚洲肠道微生物群具有大量未表征的肠道微生物遗传多样性
MAG分析表明可恢复肠道微生物90%以上的物种多样性。本研究构建的菌株级数据库可用于识别来自新加坡独人群的更多肠道细菌reads,最丰富的20种肠道细菌中,只有不到20%的菌株也被UHGG所代表,确定了27,084个生物合成基因簇(BGC)。超过90%的GCF与标准数据库中的已知BGC没有相似性,并且在肠道微生物参考基因组中的注释中未发现,展现了使用互补算法进行生物勘探的价值。